



Predicting 3D Structure Of The Hemoglobin Protein With Alphafold 2
Last Updated on August 10, 2021 by Editorial Team
Author(s): Ömer Özgür
Artificial Intelligence
Deepmind’s academic paper came out in Nature describing all the details of its CASP-winning AlphaFold v2 model for predicting protein structures. At the same time, they released all its code open source at Github.
In a short time, Google Colab notebooks were created. Thanks to this development, everybody can predict their favorite protein.
This article will see how we can use Alphafoldv2 to make some predictions and the changes taking place in the scientific world.
Biologists’ Dream
According to Levinthal’s paradox, it would take longer than the age of the universe to enumerate all the possible configurations of a typical protein before reaching the right 3D structure.
Protein is the main component of muscles, bones, organs, skin. Proteins are commonly referred to as building blocks of the human body. The function that a protein performs is largely related to the shape it is folded in. Predicting the form of a protein can solve some of the world’s biggest challenges, like developing effective treatments for diseases and finding suitable enzymes for certain tasks.
Many protein structures can be determined by experimental techniques such as X-ray or neutron diffraction, nuclear magnetic resonance (NMR), or electron cryo-microscopy (also called cryo-electron microscopy, cryo-EM). But these techniques are difficult, slow, and expensive. Also, in some types of proteins, these techniques don’t work well.
This is where AlphaFold comes to help. It’s a neural network-based algorithm that’s performed astonishingly well on the protein folding problem. In the future, we can realistically predict protein folding using algorithms like Alphafold.
Now it is AI time in biology.
Alphafold 2
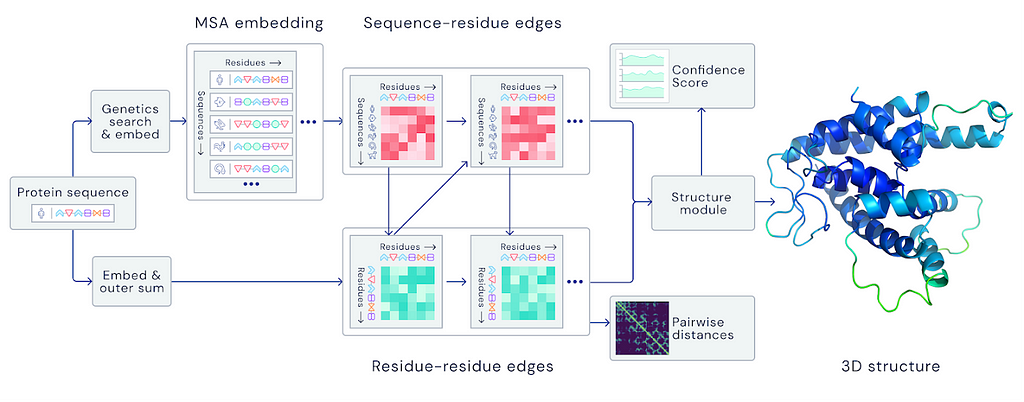
In this section, we’ll look at how Alphafold works in general and where it’s evolving.
SPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFEALERMFLSEALERMFLSFPTTKTYF
Alphafold takes the amino acid sequence at the simplest level and predicts the positions and bond angles of molecules in 3D space.
Interestingly, Alphafold uses unsupervised learning in a way similar to GPT-3.
According to the diagram in DeepMind’s blog post, MSA(Multiple Sequence Alignment) appears to be an important early step in the model. GPT-3
GPT-3 learns the general features of the language by using unlabelled text data. Similarly, Alphafold learns embedding from the sequence of proteins with similar functions.
Two protein sequences might be similar to another because the two share a similar evolutionary origin. The more similar those amino acid sequences, the more likely those proteins serve a similar purpose for the organisms they’re made in.
Some Proteins mutate and evolve, but their structures tend to remain similar despite the changes. The idea of using correlated mutations to extract structural information from an MSA is decades old and collecting pieces of other proteins to model your target’s structure.
One of the major differences between AlphaFold 1 and AlphaFold 2 is that AlphaFold 1 used CNNs, and the new version uses Transformers.
After understanding the algorithmic structure, we can start learning domain knowledge.
Hemoglobin Protein
Hemoglobin is the protein molecule in red blood cells that carries oxygen from the lungs to the body’s tissues and returns carbon dioxide from the tissues back to the lungs.
Hemoglobin also plays an important role in maintaining the shape of the red blood cells. Abnormal hemoglobin structure can, therefore, disrupt the formation of red blood cells and impede their function and flow through blood vessels.
Hemoglobin consists of protein subunits. A protein subunit is a single protein molecule that assembles with other protein molecules to form a protein complex. A process similar to making a car, the cell is producing different parts.
In some protein assemblies, one subunit may be a “catalytic subunit” that enzymatically catalyzes a reaction, whereas a “regulatory subunit” will facilitate or inhibit the activity.
In humans, hemoglobin A (the main form of hemoglobin present in adults) is coded for by HBA1, HBA2, and HBB. The hemoglobin subunit alpha 1 and alpha 2 are coded by the genes HBA1 and HBA2. The HBB gene codes subunit beta.
Getting The Sequence
Before making predictions, we need the amino acid sequence information of the protein. We can also extract the amino acid sequence from DNA using the direct amino acid sequence.
Using the NCBI database, we can access the information of a known protein and download it in fasta format.
hemoglobin subunit alpha [Homo sapiens]
Sent to → File → Format(Fasta) → Create File
What is NCBI?
The National Center for Biotechnology Information (NCBI) develops and maintains molecular and bibliographic databases as a part of the National Library of Medicine (NLM). They do not generate their own data. You can find tons of data here.
What is Fasta Format?
In bioinformatics and biochemistry, the FASTA format is a text-based format for representing either nucleotide sequences or amino acid sequences, in which nucleotides or amino acids are represented using single-letter codes.
Let’s Use Alphafold 2
- I am sharing the important code blocks here. All the codes can be accessed from Colab Notebook.
- This notebook does NOT include the alphafold2 MSA generation pipeline and is designed to work with a single sequence(no MSA).
While accuracy will be near-identical to the full AlphaFold system on many targets, a small fraction have a large drop inaccuracy due to the smaller MSA and lack of templates. For best reliability, we recommend instead using the full open source AlphaFold.
Let’s read the fasta file we downloaded and get the sequence information we need.
from Bio import SeqIO
fasta_sequences = SeqIO.parse("sequence.fasta",'fasta')
for record in fasta_sequences:
print(record)
protein_seq =str(record.seq)
- ID: NP_000508.1
- Name: NP_000508.1
- Description: NP_000508.1
- hemoglobin subunit alpha [Homo sapiens]
- Number of features: 0
- Seq(‘MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSA…KYR’)
Making Prediction
%%time
feature_dict = {
**pipeline.make_sequence_features(sequence=protein_seq,
description="none",
num_res=len(query_sequence)),
**pipeline.make_msa_features(msas=[[query_sequence]],
deletion_matrices=[[[0]*len(query_sequence)]]),
**mk_mock_template(query_sequence)}
plddts = predict_structure("alphaH",feature_dict,model_runners)
For hemoglobin subunit alpha, it took 46 min to predict. When the prediction is complete, it will be saved in the directory with alphaH_unrelaxed_model_1.pdb file.
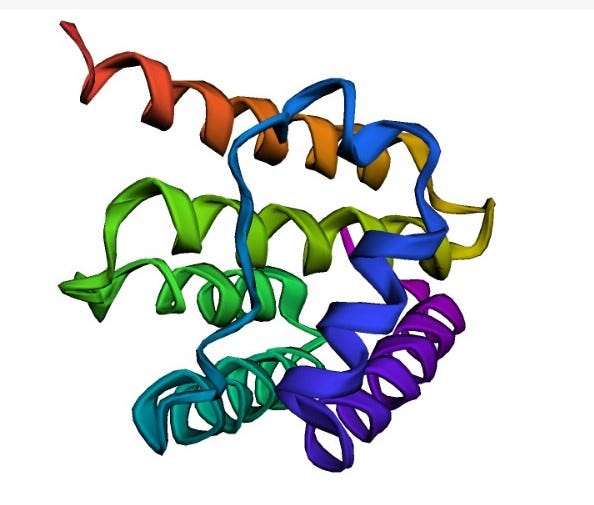
Visualization
The pdb format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, secondary structure assignments, as well as atomic connectivity.
p = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js')
p.addModel(open("alphaH_unrelaxed_model_1.pdb",'r').read(),'pdb')
p.setStyle({'cartoon': {'color':'spectrum'}})
p.zoomTo()
p.show()
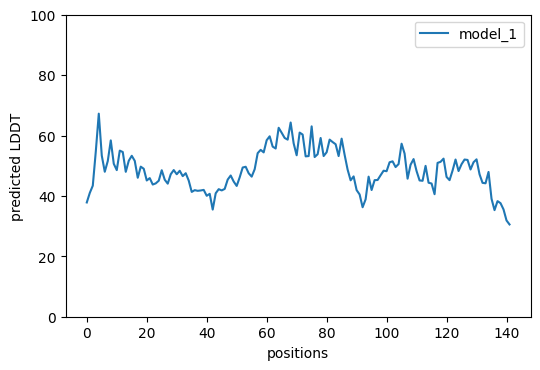
And we can visualize confidence per position. Confidence doesn’t seem too high.
Conclusion
Alphafold’s transition from academia to the open-source world could lead to many revolutions. Many scientific types of research will accelerate and very important discoveries can be made thanks to Alphafold.
One day, Alphafold may be the first AI to win a Nobel Prize.
Predicting 3D Structure Of The Hemoglobin Protein With Alphafold 2 was originally published in Towards AI on Medium, where people are continuing the conversation by highlighting and responding to this story.
Published via Towards AI
Towards AI Academy
We Build Enterprise-Grade AI. We'll Teach You to Master It Too.
15 engineers. 100,000+ students. Towards AI Academy teaches what actually survives production.
Start free — no commitment:
→ 6-Day Agentic AI Engineering Email Guide — one practical lesson per day
→ Agents Architecture Cheatsheet — 3 years of architecture decisions in 6 pages
Our courses:
→ AI Engineering Certification — 90+ lessons from project selection to deployed product. The most comprehensive practical LLM course out there.
→ Agent Engineering Course — Hands on with production agent architectures, memory, routing, and eval frameworks — built from real enterprise engagements.
→ AI for Work — Understand, evaluate, and apply AI for complex work tasks.
Note: Article content contains the views of the contributing authors and not Towards AI.
Related posts
Recent Posts
")



")


