



How To Discover Antiviral Drugs With Deep Learning?
Last Updated on May 6, 2021 by Editorial Team
Author(s): Ömer Özgür
Deep Learning
Drug discovery is a time-consuming and expensive process; deep learning can make this process faster and cheaper. Drug discovery can be divided into three parts.
- Drug properties prediction
- Drug Discovery
- Drug-target interaction prediction
Machine learning problems are broadly divided into three subgroups: supervised learning, unsupervised learning, and reinforcement learning. Drug characteristics prediction can be stated as a supervised learning problem.
Input: Molecule
Output: The degree of a chemical property (toxicity) of the molecule
Drug discovery is an unsupervised learning process.
Let’s get started
Data collection: First of all, we need information on successful antiviral drugs. We can access the active ingredients of the antiviral medications from the American Chemical Society and the medicines used to treat the MERS epidemic from Kaggle.
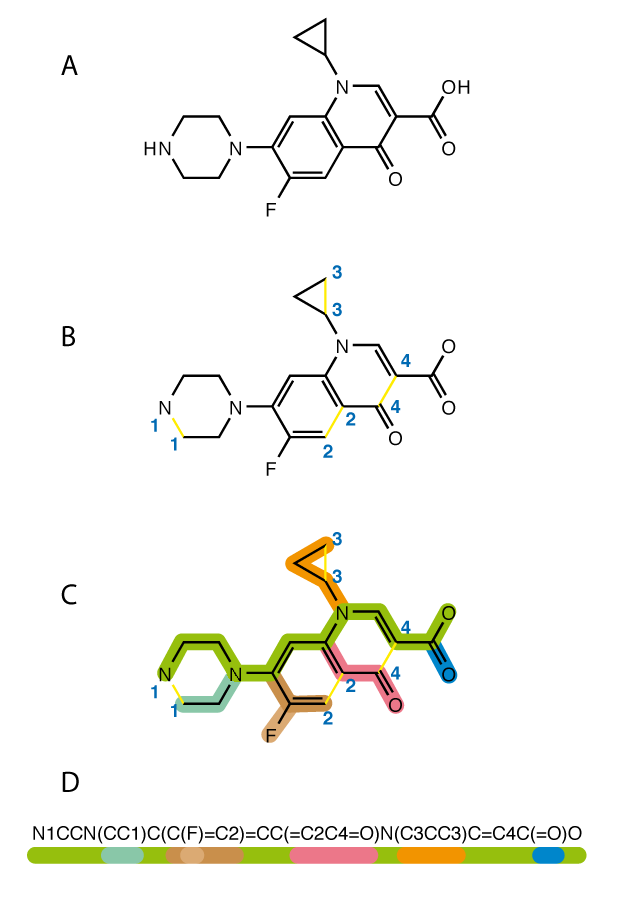
Computers do not process information that is not mathematically expressed, so we must mathematically express our molecules. The methods we can use for this:
- Molecular fingerprints
- With strings (SMILES)
- Graphic structures (2d or 3d graphics)
We will use the SMILES (Simplified molecular-input line-entry system) method to represent molecules. Briefly, we can convert a 2-dimensional molecule to a 1-dimensional text structure. It is a prevalent method and is prone to natural language processing.

Here, molecular fingerprinting takes place. Since molecules are similar to graphs, atoms form nodes, and atomic bonds form edges. We can represent nodes with vectors.
Vectors carry information such as the number of hydrogen atoms connected, the atom’s charge, the type of atom, for example (2,2, 1,3,0,1… n). At the same time, vectors are updated with information from their neighbours, thus learning geometric information.
While developing drugs, the library we use most will be DeepChem. DeepChem aims to create high-quality, open-source software for drug discovery, materials science, quantum chemistry, and biology.
Variational AutoEncoder why and how it works
Our goal with VAEs is to express the information of molecules encoded discretely in multidimensional continuously. The Neural Network, which has been trained with thousands of molecules, basically consists of 3 functions: encoder, decoder and predictor.
Mathematically representing molecules in a continuous format allows us to discover new drugs; we select random vectors from the molecular space and translate them into the molecular structure with the decoder.
When we examine the architecture of AutoEncoders, we encounter three structures:
1- Encoder: Here, the artificial neural network learns how to represent information in a smaller size. Briefly, the data starts to get compressed as it moves towards the bottleneck and essential functions are removed; it can also be thought of as nonlinear dimension reduction.
2- Bottleneck: When the structure of the AutoEncoder is examined, it is actually like a butterfly; the bottleneck is the part where the information passes between the wings. Data is represented in a compressed multidimensional vector space.
3- Decoder: Learns how to reconstruct the encoded representation as close as possible to the original input.
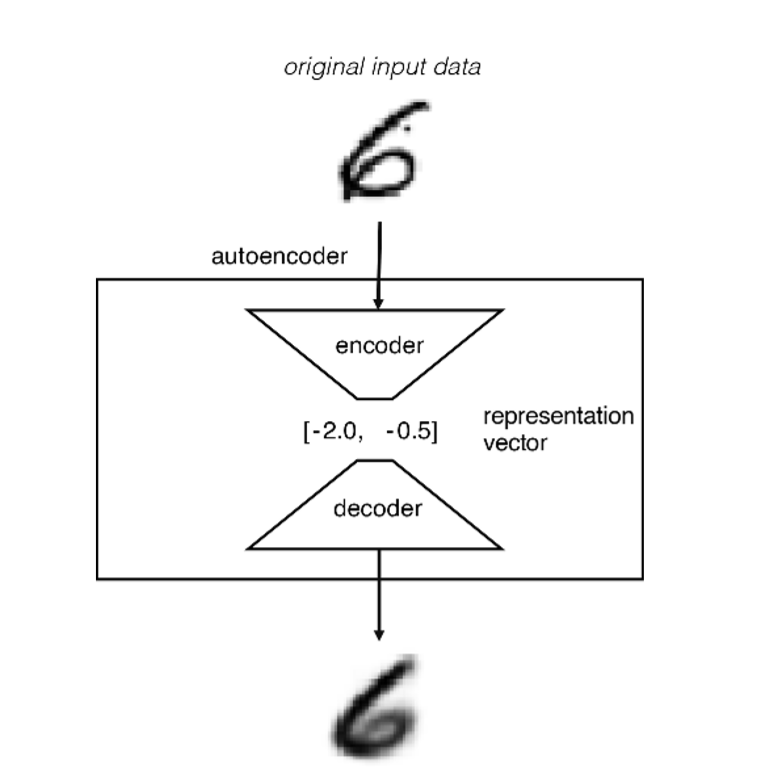
As in every Neural Network, we need to create or define a cost function. We know that this method is an unsupervised learning process, and we want it to reconstruct the SMILES format of the molecule.
The cost function we describe measures the difference between input and output. How successfully it can generate the input. If we are to input and output an image, we try to minimize the difference between the numbers representing the colour intensity in its pixels.
Difference of the Variational Autoencoder
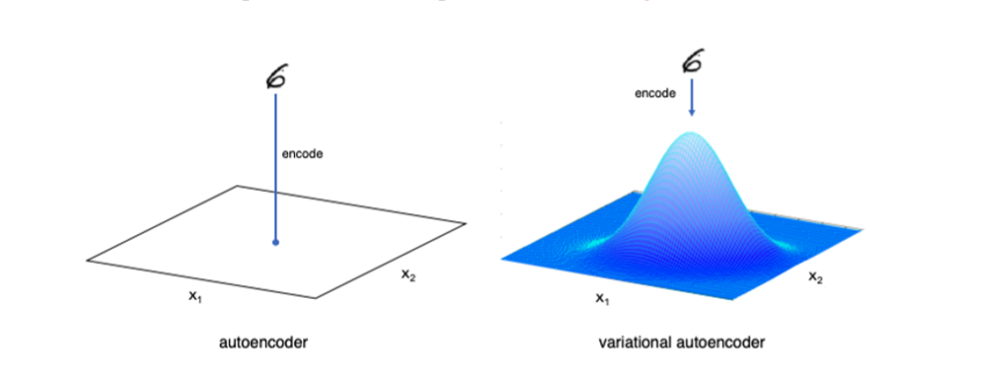
While autoencoders reduce the input to a single point in space, Variational autoencoders force the encoded vector space to disperse with Gaussian distribution (Normal Distribution).
As a result, we can achieve better quality feature extraction. It performs a different measurement between the probability distribution. When loss function gets smaller, the closer the vector space to the normal distribution.
Generative adversarial networks are used to generate things that did not exist before. At the same time, it makes more sense to use VAE to create different types of the same kind of information (drug, image, sound).
Artificial neural network architectures used in drug discovery
Character strings (SMILES) can be encoded into vectors using recurrent neural networks (RNNs). Sequential learning can be performed using 1D CNN or RNN in the encoder part and RNN in the decoder. RNN or GRU in the last layer predicts which characters should be where.
Drug Discovery
Drug discovery is a bit like looking for a needle in a haystack. However, the drugs we create can be expressed mathematically, the laws of chemistry and physics may not allow when you want to synthesize them, so the pool of molecules that started in large numbers shrinks as they go through the elimination stages, some of these stages are:
- If the number of atoms of the molecule is less than 10, there is not enough interaction energy to react.
- If the molecule has more than 50 atoms, it is difficult to dissolve in water, and problems can occur in biological reactions (in general).
QED(Quantitative estimate of drug-likeness)
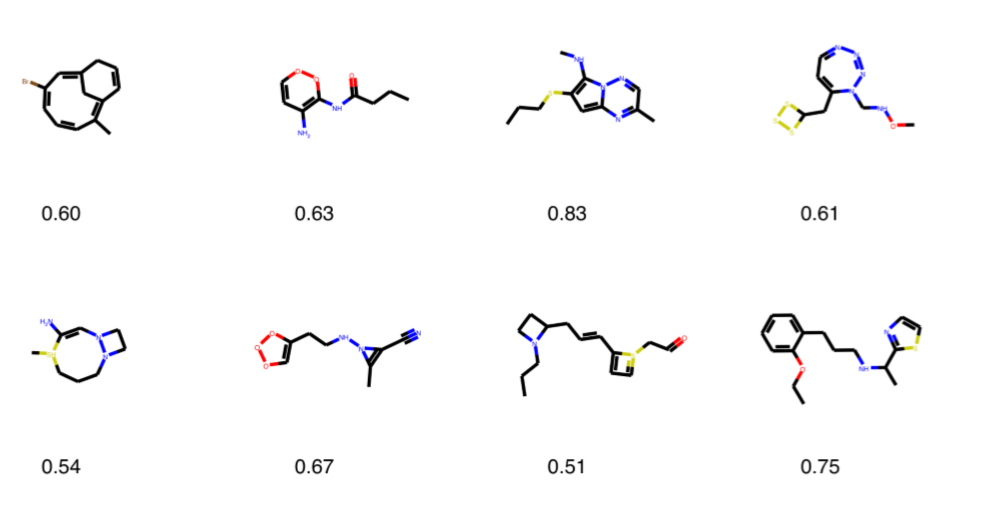
It is the method by which we measure the potential of a molecule to become a drug. We can do this using the RDkit library; each molecule is scored between 0–1, we can pass those above 0.5 points to the next stage.
The most successful molecule ever discovered in this experiment
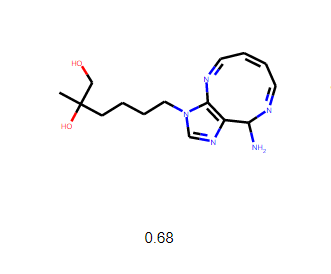
The most successful molecule developed, the QED score of 0.68
To measure the molecule’s biological activity, a chemist must first be synthesized, and then we can pass the test stages. As a result, drug discovery is a new field and is evolving. It aims to facilitate long processes involving trial and error using human intuition.
— Resources —
How To Discover Antiviral Drugs With Deep Learning? was originally published in Towards AI on Medium, where people are continuing the conversation by highlighting and responding to this story.
Published via Towards AI
Towards AI Academy
We Build Enterprise-Grade AI. We'll Teach You to Master It Too.
15 engineers. 100,000+ students. Towards AI Academy teaches what actually survives production.
Start free — no commitment:
→ 6-Day Agentic AI Engineering Email Guide — one practical lesson per day
→ Agents Architecture Cheatsheet — 3 years of architecture decisions in 6 pages
Our courses:
→ AI Engineering Certification — 90+ lessons from project selection to deployed product. The most comprehensive practical LLM course out there.
→ Agent Engineering Course — Hands on with production agent architectures, memory, routing, and eval frameworks — built from real enterprise engagements.
→ AI for Work — Understand, evaluate, and apply AI for complex work tasks.
Note: Article content contains the views of the contributing authors and not Towards AI.
Related posts
Recent Posts




")

